This walk through shows how to use additional
diagnostic tools, specifically the nearest lines window and
intensities to aid the automatic assignment process on the N2O
described in the previous section. These additional diagnostic
tools are not essential for N2O, but are very helpful
in more complicated cases.
-
For the purposes of this walk through the
overlay (
N2Onu2.ovr)
and line list (
N2Onu2lin.ovr)
from the previous example can be used.
-
As previously, an approximate simulation is
required. The same set up as before can be used - "File",
"New", "Linear molecule", set the excited
state Lambda = Pi, B"= 0.45, B'
= 0.451, Origin = 588.7 cm-1.
-
At this stage a different approach is used
to select the trial fit and check transitions. As before the P
and R branches are used, but all are added to the working line
list:
-
Open the transitions window, and select
the P and R branch lines by setting "Change" to "<>".
-
Use the Add button to add all
these lines to the line window.
-
For subsequent operations, it is helpful
for these to be sorted by J. In the line list
window, select "More", "Sort On", "Upper
State".
-
In the line list window, select "More,
Advanced" to make the autofit options visible.
-
The nearest lines window will now give
useful results. Bring up this window with "More", "Plot
Nearest Lines". At its simplest this window plots, for
each line in the line list window, a point for each observed
line within the given Y range. If the simulation is perfect,
than a clear horizontal line should appear along the centre
zero line, together with a typically random set of points
either side. In the current case (while the simulation is not
that close) the default Y Range is too small - try 1
for this. Possible assignments are indicated by curved line,
but we will not make use of them here.
-
To make use of intensity information, we
require peak intensities to be added to the line list as part
of the assignment process, as well as positions. To do this,
make sure "Set I" is selected in the line list
window. This will display another column, Isigma,
which works as for the Std Dev column, but for the
intensities.
- Set up the autofit as follows:
- Select a range of J, say J' = 10-15 in the line list
window by clicking and dragging.
- Mark these as check transitions by right clicking and
selecting "Mark as Check". A bold C
will appear in the "Std Dev" column, and open triangles will
indicate their position in the main plot window.
- Select three transitions as trial fit transitions, say the
first and last, and one in the middle by click on the
individual lines, right clicking and selecting "Mark for
Autofit". A bold F will appear in the
"Std Dev" column, and filled triangles will indicate their
position in the main plot window.
- Decide on the controlling parameters for the autofit. There
are two key settings:
-
The acceptance window, which is the
maximum error expected for the check transitions after the
trial fit. This is set in the box labelled "Accept"
in the line list window. For the purposes of this
walk-through we will leave this at the default (0.1), though
in general a number of the order of the line width
(here 0.001) is more appropriate.
-
The search window, which controls how far
each side of the initial "fit" line positions you want to
search. The intent is that the approximate initial
simulation should predict the "fit" transitions to be out by
no more than this value. For this example 3 cm-1
might be a good value. The search time will increase rapidly
as this is increased, so in more complex cases the range may
need to be considered carefully. It is set in the autofit
window, which is brought up with "Overlays, Autofit...".
The Test button in this autofit window will
display the number of trials in the box at the top, and
display some additional information in the log window.
- We are now in a position to try an autofit. The file saved at
this stage (before the autofit) is available as N2OautoD1.pgo.
Using the automatic fit results is as in the simplified version -
double click on a trial to update the constants, assignments and
plots for that trial. Note the following additional points:
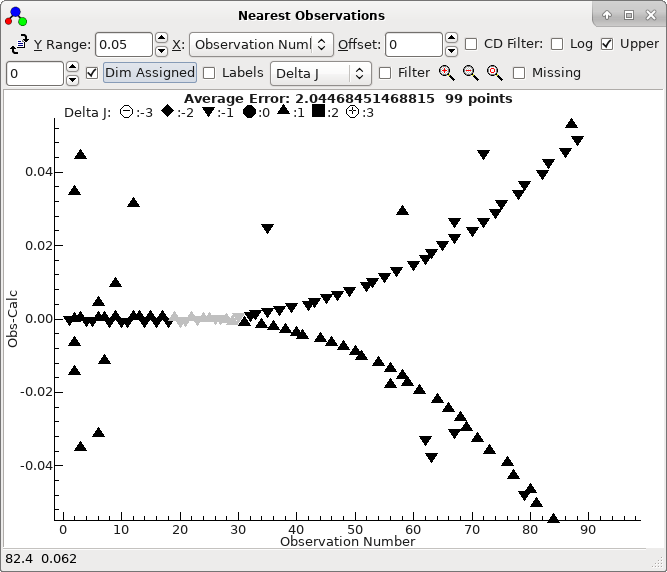
The nearest lines window comes into its own at this stage, as
assignments can be made directly from it, and it is typically easy
to see the correct assignments. In the current case the plot looks
like this:
To obtain this exact plot "
Dim Assigned"
has been selected which plots the assigned points in grey. Recall
this window plots, for each transition in the line list window,
the distance to all observed lines within a particular window,
here 0.05 cm
-1. It is clear from this plot that most,
if not all, of the unassigned lines have a transition within this
window, and the obvious assignments to take are the ones that lie
on the smooth curves. In this case the density of transitions is
such that this is the point closest to zero in each case, though
is the density of lines was higher the smooth trend can be easily
picked out. The two curves correspond to the P and R branch
transitions. Assignments can be from this window as follows:
- To assign the low observation numbers, draw a box with the
mouse to enclose the observations lying on the horizontal
lines to the left, right click, and select "Assign Points
Inside":
- To assign along the curved lines, click and drag with the
mouse so that the diagonal line plotted lies along the line,
right click and select "Assign On Diagonal":
This will assign points close to the line.
- Repeat as required; if you make a mistake the various "Clear
Point..." actions are available by right clicking, and work in
the same way as the residuals window. (The residuals window
will be automatically updated, and it is also possible to fix
problems there.)
- Fit as before, adjusting assignments as required. At this
stage the centrifugal distortion constants must be floated.
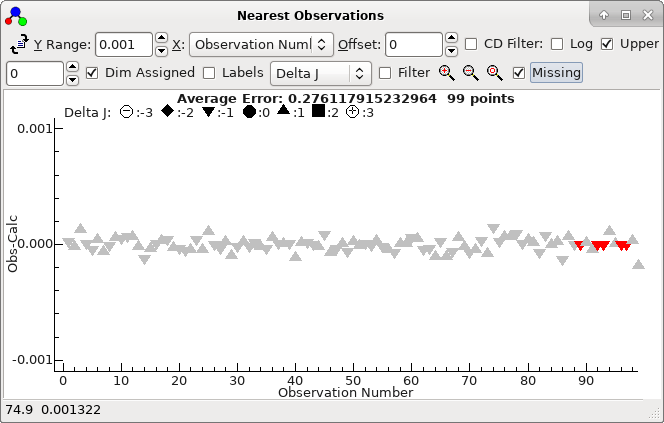
After a couple of cycles of assignment and fitting the nearest
lines window must be zoomed in much further (by a factor of
50) to show the quality of the fit:
This indicates there are no more obvious assignments that can
be made. The "Missing" option has been selected here,
plots a point (at zero) for each line that has no observation
in the selected window. (As you zoom out these will disappear
as a transition moves in range.) To investigate consider one
or more of the following:
- Right click on one of them and select "Show and Edit" to
centre the main plot on the predicted position
- Changing the x axis in the nearest lines window
to "Frequency".
In this case either of these will show that missing lines are
outside the range of the experimental spectrum.
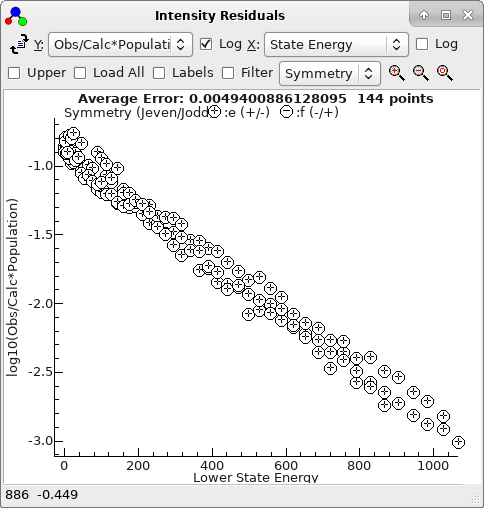
The intensity window is also giving useful information at this
stage. If not already visible bring it up with "
View", "
Intensity
Residuals...", and it should look something like this:
By default, this is a plot of the log of the ratio observed
intensity to calculated line strength, i.e. notionally log
10(observed
population) and plotted against lower state energy should give a
linear Boltzmann plot, and here gives additional confirmation that
the assignments are plausible. Note that this is choice of plot is
independent of the temperature chosen for the simulation. This
window has the same tools for adjusting assignments as the main
residuals window. A good application is to reject transitions that
have much too high an observed intensity (that appear above the
trend line) but with correct positions. This implies a weak
component of a blend has has been assigned, which should probably
not be included in a fit.
The intensity window above is not indicating any problems, so the
assignment of the P and R branches is now complete; The file at
this stage is saved as
N2OautoD3.pgo.
D. Determining additional parameter(s) - the Q branch
We now assign the Q branch lines:
-
Open the transitions window, and select
the Q branch lines by setting "Change" to "Q".
-
Use the Add button to add all
these lines to the line window.
- Select (say) the first 10 Q branch lines as check
transitions, and one (say Q(4)) as a fit transtion.
- In the constants window, float q in the upper state.
-
Make sure all all the standard deviations
are blank (click on the down arrow in the autofit window and
select "Clear Parameter Ranges") otherwise they
will act as constraints on the search.
- A much smaller search window is needed - 0.2 cm-1
should be plenty.
The file set up for the autofit is saved as
N2OautoD4.pgo. In this case the
top ranked trial is correct, and the same process above allows for
rapid assignment of the Q branch lines. The nearest lines window
is again invaluable. To show only the Q branch lines in this
window (and exclude the already dealt with P and R branch lines)
select "
Filter". When this option is enabled, any
selection set up in the transitions window is also applied to the
nearest lines window. The final file, after floating
qD
is saved as
N2OautoD5.pgo.
In this section, the intensities, rather than the peak positions
are fitted, giving a value for temperature.
 Procedures Automatic Fitting
Procedures Automatic Fitting