Molecule Types Linear Molecules
Molecule Types Linear Molecules | Molecule Types Linear Molecules |
<Prev Next> |
In this example we will go through the process of analysing the
spectrum shown below which is a fragment of the ultraviolet LIF
spectrum of C3. This will highlight some of the
considerations that go into assigning and simulating a spectrum.
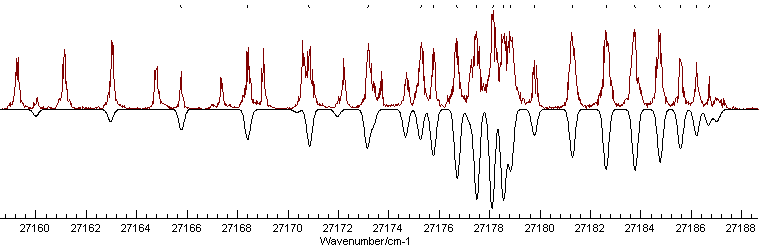
This spectrum is available as C3AX.ovr. The near UV electronic spectrum of C3 is known to be dominated by an A1Πu - X1Σ+g transition, and a rotational constant of ~0.5cm-1 is typical for a molecule of this size. This gives us enough information to set up a basic data file, as summarized in Making a Linear Molecule Data File; remembering to set AsymWt = 0 as there are two equivalent spin zero nuclei. The spectrum is taken in a molecular beam, which normally gives very low rotational temperatures, so try 30 K for the temperature. Setting this up, with an origin of 27178 cm-1 should give something like this:
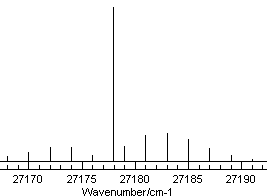
This shows a strong Q branch, which is clearly
present in the experimental spectrum at around 27178 cm-1
and the branch to the right looks as though it could be assigned -
note the small gap immediately to the right of the Q branch, which
is also visible in the spectrum. Using the Line Position Fitting procedure to
assign the first few members of this branch, starting from the
left gives the picture on the left on pressing "Test":
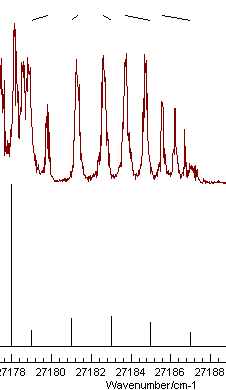 |
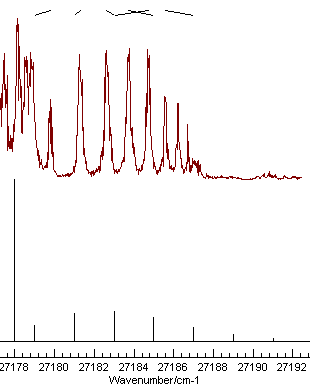 |
| Good |
Mis-assignment |
Note the nice pattern of indicating marks at the top for the spectrum on the left, joining the positions of the observed and calculated line positions. The plot on the right shows a typical plot following a miss-assignment.
We can now try some fitting; it normally makes sense to start by fitting just a few parameters, so let's try the upper state origin and B:
5 Observations, 2 Parameters
Initial Average Error: 1.22114672883084
Predicted New Error: 0.34497794153667
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 27178 27178.5648453395 .2402865 2.3507 .017249 27178.56(24) A v=1 Origin
2 .5 .474483017372344 .0048478 -5.2636 .000348 0.4745(48) A v=1 B
These two parameters are well determined, and the plot shows a
much better fit:
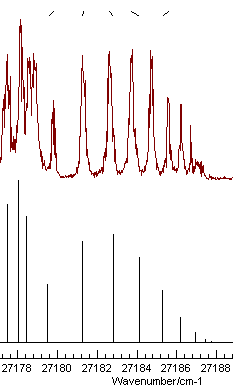
It also indicates one of the lines has been
miss-assigned - the rightmost one. Note that holding the mouse
over the tick mark at the top will show the source of the relevant
observation, and right clicking will take you to the line in the
line-list window for editing. Correcting the error and re-fitting
gives an even better fit. The obvious additional parameter to
float is the lower state B value, and the fit with these
observations does indeed work:
5 Observations, 3 Parameters
Initial Average Error: 0.0229832236953073
Predicted New Error: 0.0229832236953073
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 .431694643119403 .431694642718755 .00863921 0.0000 1.99e-5 0.43169(864) X v=0 B
2 27178.9571892846 27178.9571892862 .033426 0.0000 .000766 27178.957(33) A v=1 Origin
3 .409391071646742 .409391071313973 .00714522 0.0000 1.55e-5 0.40939(715) A v=1 B
Correlation Matrix
1 2 3
1 1.000
2 -0.878 1.000
3 0.999 -0.894 1.000
While the values are physically reasonable, note the large correlation of 0.999 between parameters 1 and 3, the two rotational constants, which is also reflected in the estimated errors for these parameters being quoted to 3, rather than 2 figures. This implies that there is only just enough information in the supplied data to determine the three parameters, and the derived values should be looked at with some suspicion.
The correlation can be broken by adding more
lines, preferably from another branch. Looking at a wider range of
the spectrum indicates several possible extra assignments, though
there are clearly more lines in the experimental spectrum than the
simulation, indicative of an additional band being present:
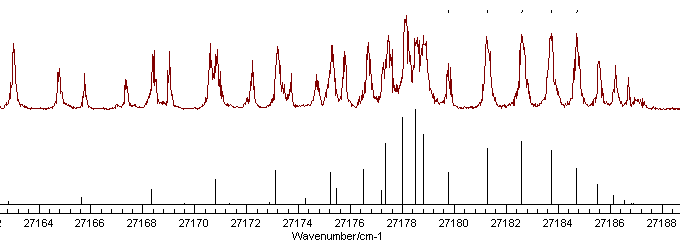
At this stage it is probably simplest to add
several assignments, and then look for any that don't look quite
right. Assigning 14 more lines that look about right leads to an
improved fit:
19 Observations, 3 Parameters
Initial Average Error: 0.0585913375363875
Predicted New Error: 0.0585913375363875
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 .429544716011829 .42954471601421 .00101655 0.0000 2.03e-5 0.4295(10) X v=0 B
2 27178.9692155569 27178.969215557 .02145922 0.0000 .001953 27178.969(21) A v=1 Origin
3 .408140469786951 .408140469789101 .00094343 0.0000 1.93e-5 0.40814(94) A v=1 B
Correlation Matrix
1 2 3
1 1.000
2 0.227 1.000
3 0.976 0.058 1.000
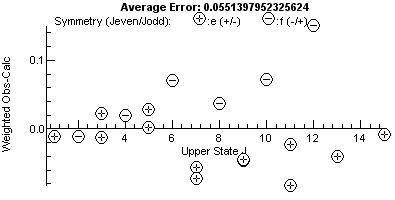
Note how the correlation between the the
rotational constants (parameters 1 and 3) is no longer 0.999. At
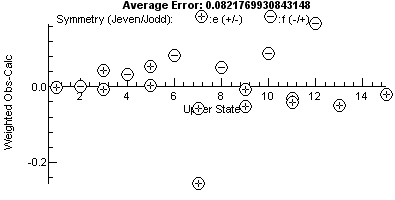
this stage the residuals window (View, Residuals) is perhaps the
best way of looking for problems; compare the fit on the left with
the one on the right where one assignment has been shifted to the
other peak of a doublet (the pair at 27170.5 cm-1):
|
|
|
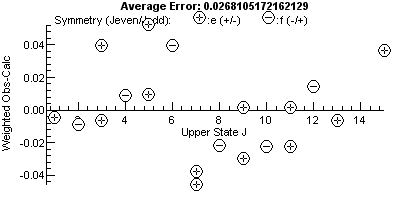
Note the plot on the right has a bigger vertical
scale, and one point, the miss-assigned peak, stands out. For a
good fit, the residual fit should look completely random, and a
useful trick to indicate issues is to try different horizontal
scales. The plots above are against upper state J, and the
errors are clearly bigger at higher J. This might be a cue
to try centrifugal distortion (D), though in this case note
that the f parity levels (minus signs in circles) are
above the axis and the e (+) levels are below the axis.
This is a classic symptom of lambda doubling, and allowing q
in the upper state to float gives a much improved fit, and the
residual plot now has no obvious trend:
19 Observations, 4 Parameters
Initial Average Error: 0.0301742415918127
Predicted New Error: 0.0301742415918126
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 .430438760425889 .43043876044772 .00053537 0.0000 7.85e-6 0.43044(54) X v=0 B
2 27178.9607318607 27178.9607318605 .01110167 0.0000 .000754 27178.961(11) A v=1 Origin
3 .409363044507011 .409363044540297 .0005115 0.0000 7.47e-6 0.40936(51) A v=1 B
4 -.00117830836843453 -.00117830840808154 .00016929 0.0000 1.49e-5 -1.18(17)e-3 A v=1 q
Correlation Matrix
1 2 3 4
1 1.000
2 0.201 1.000
3 0.972 0.025 1.000
4 -0.209 0.095 -0.313 1.000
If you don't float the right parameters there are
various possible indications. If you float p (the other
commonly used lambda doubling parameter) instead of q
then PGOPHER clearly detects this is not determined:
19 Observations, 4 Parameters (scaled)
Initial Average Error: 0.0620577920690341
Predicted New Error: 0.0620577920690341
**** 1 combinations of parameters not floated **** (SVD threshold = 1E-6)
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 .429658997291519 .429658997292251 .00107669 0.0000 1.61e-5 0.4297(11) X v=0 B
2 27178.9680780115 27178.9680780115 .02272882 0.0000 .001551 27178.968(23) A v=1 Origin
3 .408250076142484 .408250076143445 .00099924 0.0000 1.54e-5 0.40825(100) A v=1 B
4 0 0 NAN NAN INF 0 NAN A v=1 p
Correlation Matrix
1 2 3 4
1 1.000
2 0.227 1.000
3 0.976 0.058 1.000
4 Nan Nan Nan Nan
Singular Value Decomposition
X v=0 B A v=1 Origin A v=1 B A v=1 p
1 606.33631228 -.68905037825 .005563083443 .724692092088 0
2 42.9863049189 .724633451607 -.00956828901 .689068072586 0
3 2.73019060661 .010767406567 .999938748098 .002561836805 0
4 0 0 0 0 1
This case is particularly clear, in that the singular value decomposition has indicated exactly which parameter is not determined - the rows in the singular value decomposition correspond to linear combinations of parameters, the the lowest row indicates the combination that is least well determined. In this case it is clearly p, and indeed inspection of the Hamiltonian will indicate that p requires non zero spin. A similar result will be obtained if you try and float p or q for the ground state, as these parameters are not relevant for Σ states.
Floating D in either state gives a
rather better behaved fit, though the error bar is larger than
the parameter, indicating it should be fixed at zero:
4 -3.3058739831523e-7 -3.3059827872211e-7 3.3351e-6 0.0000 8.77e-8 -3(33)e-7 A v=1 D
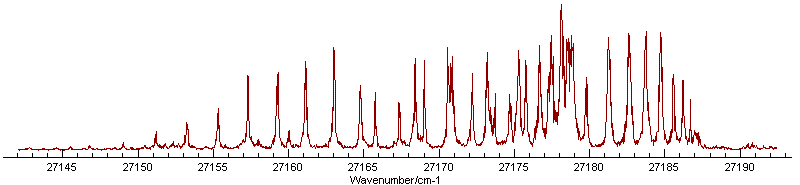
Adjusting the temperature to 50 K and the
(Gaussian) linewidth to 0.25 cm-1 gives an excellent simulation
of most of the spectrum, though there is clearly an additional
band present: