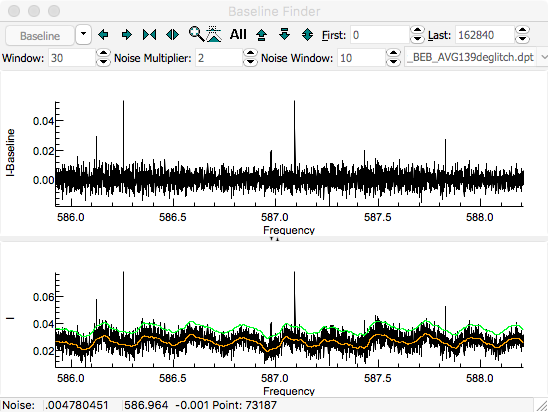
Press the "
Baseline" button to
calculate a baseline. The orange line shows the calculated
baseline, and the green line indicates the upper limit of the
points used in calculating the baseline. The upper pane shows
the spectrum after subtracting the baseline. The expand
vertical button (

) can be helpful in expanding the baseline.
Experiment with the "
Window", "
Noise Multiplier",
and possibly the "
Noise Window" settings and with "
Dense"
selected from the drop down menu. (Click on the down arrow
next to the "
Baseline" button to see this.) You need
to click on the "
Baseline" button after changing the
settings to update the display. The baseline algorithm works
by attempting to identify points on the baseline (within the
noise multiplier) and taking a moving average over the window.
A "
Window" of 30, "
Noise Multiplier" of 2
and selecting "
Dense" gives good results for this
spectrum:
 Procedures Automatic Fitting
Procedures Automatic Fitting