 Molecule Types Vibrational Structure
Molecule Types Vibrational Structure | Molecule Types Vibrational Structure |
<Prev Next> |
The force field can be expressed in terms of valence or
symmetry coordinates. A typical analysis will include several
isotopologues and might have a data file structured something
like this:
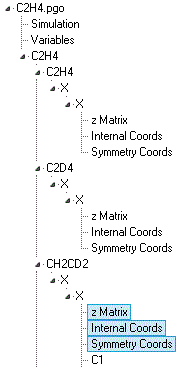
Note the various single and multiple objects:
A Cartesian Coordinates
object may also be present; the exact combination of objects
used depends on the required calculation. Nuclear Co-ordinates and Vibrational Modes objects will
also be present, automatically created and updated from the
force field analysis objects listed above.
The current version of the program does not explicitly make use of symmetry for the force field analysis, but it is implicit in the way the force constants and geometry are specified. The current version of PGOPHER is restricted to harmonic terms, but the program is structured so that anhamonic terms can be added in future. The units are currently less flexible than for rotational calculations; the overall mixture units must be set to cm-1, the geometry specified in Angstroms and degrees and force constants (for stretches) in millidyne/Å = aJ/Å2 (1 millidyne/Å = 1 aJ/Å2 = 100 J/m2 = 100 N/m). For a full discussion of the units see Hedberg and Mills (1993).