Molecule Types Vibrational Structure Force Field Analysis
Molecule Types Vibrational Structure Force Field Analysis |
Molecule Types Vibrational Structure Force Field Analysis |
<Prev Next> |
Symmetry coordinates, S, are specified by giving the
transformation matrix, U, between S and the
internal coordinates, R:
S = U R = U B Δx
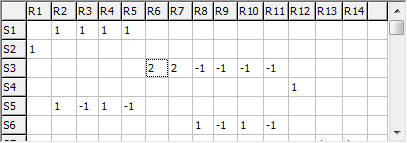
The symmetry coordinates window, shown below for C2H4, allows the display and editing of the U matrix and (optionally) the force field in terms of symmetry coordinates, F. To create a symmetry coordinates object, right click on the Electronic State object object and select "Add New..., Symmetry Coords". To bring up the window right click on the symmetry coordinates object and select "View...". If a symmetry coordinates object is not present, or the U matrix is left blank, an identity matrix is assumed for the U matrix. Note that the current version of the program ignores the point group set for the molecule as a whole, and the symmetry set for the individual vibrational modes when doing the force field calculation. This may be changed in future versions of the program.

The plot at the bottom shows the current symmetry coordinate, i.e. the selected row in the top left grid. The arrows indicate how far each atom moves. The plot updates semi-automatically; use "Operate, Check" to force an update.

| Check |
Check the contents of the top grids, and
update the plot to match |
| Apply |
Check the contents of the top grids, and
display the details of the vibrational mode calculation in
the Log Window. This also forces
other objects to update. |
| Show F Matrix | Print the F matrix in the Log
Window calculated from the l matrix
elements and frequencies given in the vibrational mode objects.
The transformation is done by calculating the matrix L,
which relates the symmetry coordinates, S, and
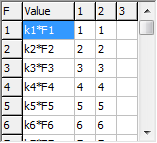
normal coordinates, Q:S = L QThe L matrix is available from: L = U B M-½ lproviding the internal coordinates are fully specified and the F matrix can then be calculated from: F = (L-1)T λ L-1Note that the U matrix is not necessarily required, but if there are redundant internal coordinates L-1 can't be calculated. In this case an alternative relationship can be used: F = L λ LTprovided that L LT= 1. |
| Set F Matrix | Clear the f matrix grid (top right) and set it from the l matrix elements and frequencies given in the vibrational mode objects. See the entry above for details of the calculation. |
| Sort F Matrix by Value | Sort the F matrix grid (top right) in descending order of the first column. |
| Sort F Matrix by Mode | Sort the F matrix grid (top right) by mode numbers, with diagonal elements first. |
V = ½ ST F S = ½ Σij FijSiSj