Automatic Fitting of an N2O IR Spectrum
This walk through shows how to use the automatic
assignment process on the N2O sample file provided in
the Walk-through of Simulating and Fitting a
Simple Spectrum (which this assumes you are familiar with).
This is perhaps too simple an example to use automatic fitting on,
as the assignment is fairly obvious, but it does demonstrate the
procedure. The process described here is general, and could in
principle work on any type of molecule or spectrum that PGOPHER
could simulate. See also the following walk through, which uses
the enhanced diagnostic tools that will not necessarily be useful
in all circumstances. Note also that the many of the choices of
peaks and values made below are arbitrary, and the walk through
could have worked just as well with many other choices.
A. Generating the peak list
- Load the overlay N2Onu2.ovr.
- Right click on the overlay and select "Baseline and
Peaks...".
-
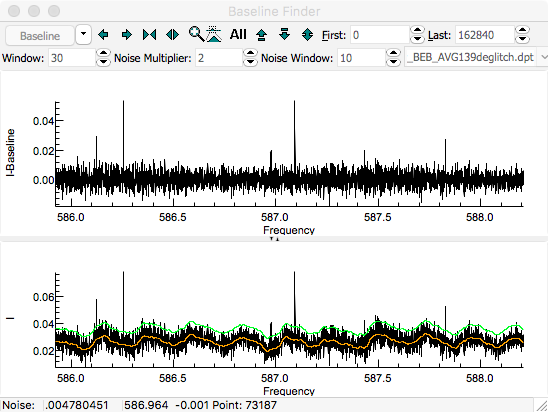
The first step is to determine a baseline.
For this spectrum, the baseline is essentially flat, but has a
small ripple, so use the zoom buttons to zoom in on a small
sample of the spectrum to show the ripple. (The zoom buttons
in this window work in the same way as the in the main
window.)
-
Press the "
Smooth" button to
calculate a baseline. The orange line shows the calculated
baseline, and the green line indicates the upper limit of the
points used in calculating the baseline. The upper pane shows
the spectrum after subtracting the baseline. The expand
vertical button (

) can be helpful in expanding the baseline.
Experiment with the "
Window", "
Noise Multiplier",
and possibly the "
Noise Window" settings and with "
Dense"
selected . You need to click on the "
Smooth" button
after changing the settings to update the display. The
algorithm used by the "
Smooth" button works by
attempting to identify points on the baseline (within the
noise multiplier) and taking a moving average over the window.
A "
Window" of 30, "
Noise Multiplier" of 2
and selecting "
Dense" gives good results for this
spectrum:
-
If you want to save the spectrum with the
baseline subtracted, select "Make New" under "Make
Overlay" to generate an overlay with a flat baseline.
-
To make a line list, adjust the "Noise
Multiplier" so that the green line is above almost all
of the noise. It is not necessarily the same as used for the
baseline calculation - in this case a "Noise Multiplier"
of 3 is promising. Selecting "Live update" will show
the lines found in the upper window in blue automatically as
the parameters are changed, though note this can be slow if
the selected region is large. A minimum width and area can be
selected; for the current spectrum the "Min Width" is
too large, so deselect it, or change the value.
-
When you are happy with the result,
generate the line list by pressing "Peak Find" and
then "Make New" under "Make Line List". The
resulting line list will be added to the overlays, and show in
the main window.
-
In the current example, the above settings
give bad results for one or two regions where the noise is
larger, so re-run the peak finder over smaller regions as
follows:
-
Select the region to update by either:
-
Zoom to the region in the
baseline window
-
Make this the active region for
peak finding pressing "Visible Here" on the
right.
or:
-
Select a region in the main window, and
zoom the main window to that region
-
Make this the active region for peak
finding by pressing "Visible in Main" in the
baseline window.
-
Adjust the noise multiplier (or other
setting) as required.
-
To replace the peaks in the line list,
press "Peak Find" then "Append" under "Make
Line List", making sure the generated line list is
selected and "Erase Current Region" is checked when
prompted.
-
Repeat as required - to reset the window,
press the "All" button in the right (not the top)
of the baseline window.
-
To delete individual lines from the line
list in the main window, right click on the line and select "Delete
Point"
-
To delete several adjacent lines, select
them (left click and drag) and then right click and select "Delete
Points".
-
To add individual lines by manual
measurement, right click and drag over the peak in the
baseline window.
-
The original experimental spectrum is not
required at this point - you may want to hide or remove it
from the main window by right clicking on it and selecting the
appropriate option. You may want to use it later, though, for
checking for weak peaks that the automatic peak finder may
have missed.
- The line list is saved as N2Onu2lin.ovr,
using "Save Overlay As...".
B. Basic Automatic Fit Operation
To start with, we set up a minimum automatic fit to determine the
origin, and upper and lower state rotational constants, by fitting
to the P and R branches. This requires the following steps:
1. Setting up the automatic fit
-
Set up an approximate simulation, and
select the parameters to be determined.
-
Generate the basic molecule type with "File",
"New", "Linear molecule" and set the
excited state Lambda = Pi.
-
Manually adjust the origin, and upper and
lower state rotational constants to produce something that
is qualitatively correct. The simulation does not have to be
that good - see the discussion of the search window below.
The description below uses B"= 0.45, B' =
0.451, excited state Origin = 588.7 cm-1.
-
The parameters to be determined, here
origin, B' and B" should have "Float"
= yes. At this stage the absolute minimum of parameters
should be floated.
-
Select the transitions for trial
assignments and fitting, the "fit" transitions. These need to
satisfy the following criteria:
-
The number should match the number of
parameters marked as floating. (An excess number can be
given, but this will dramatically increase the search
time.)
-
The transitions selected should be
sufficient to determine the parameters floated
-
The transitions should be clear in
the experimental spectrum. This necessarily can't be
known at the start of the process (as the assignments
are not known), and is one of the reasons it might well
be necessary to try more than one starting point. In
general the simulation can indicate the strongest lines
that are clear of other lines. In the current case the
simulation suggests the central Q branch region should
be avoided.
For this case three transitions are required:
-
Right click on (say) one P and two R
branch lines. (Selecting three lines in one branch will give
a poor determination of the difference between the two
constants.) The three transitions will appear in the line
list window.
-
In the line list window, select "More,
Advanced" to make the autofit options visible.
-
Click and drag the mouse to select the
three transitions, right click and select "Mark for
Autofit". A bold F will appear in
the "Std Dev" column, and filled triangles will indicate
their position in the main plot window.
-
Select the check transitions. The choice of
these is less critical than the fit transitions, as not all of
them need to be assigned as part of the autofit, but they
again should be transitions with a good chance of appearing
clearly in the experimental spectrum. In the current case a
few P and R branch transitions would be appropriate
-
Right click on (say) two R and two
P branch transitions, so they appear in the line list
window.
-
Click and drag in the line list window to
select these transitions, right click and select "Mark
as Check". A bold C will appear in
the "Std Dev" column, and open triangles will indicate their
position in the main plot window.
- At this stage, your desktop should look something like this:
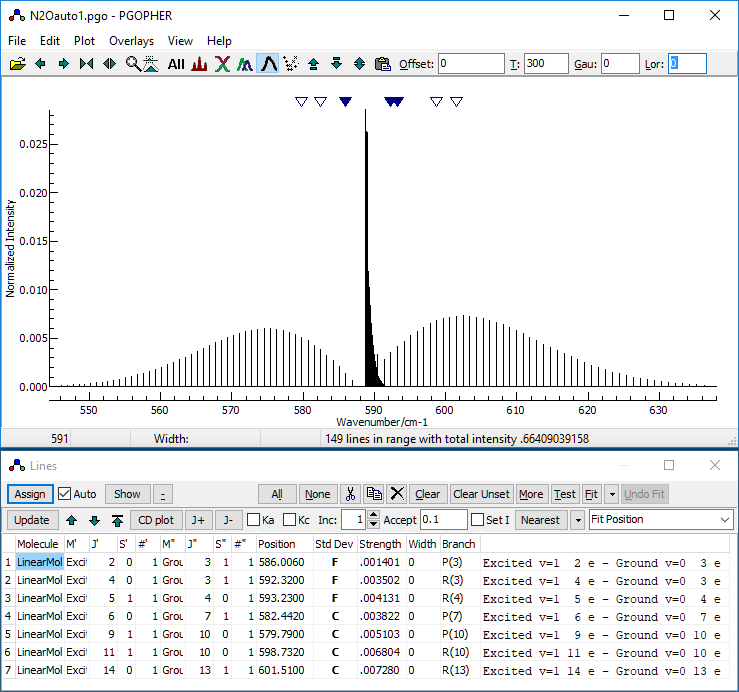
- Decide on the controlling parameters for the autofit. There
are two key settings:
-
The acceptance window, which is the
maximum error expected for the check transitions after the
trial fit. This is set in the box labelled "Accept"
in the line list window. For the purposes of this
walk-through we will leave this at the default (0.1), though
in general a number of the order of the line width
(here 0.001) is more appropriate.
-
The search window, which controls how far
each side of the initial "fit" line positions you want to
search. The intent is that the approximate initial
simulation should predict the "fit" transitions to be out by
no more than this value. For this example 3 cm-1
might be a good value. The search time will increase rapidly
as this is increased, so in more complex cases the range may
need to be considered carefully. It is set in the autofit
window, which is brought up with "Overlays, Autofit...".
The Test button in this autofit window will
display the number of trials in the box at the top, and
display some additional information in the log window.
- We are now in a position to try an autofit. The file saved at
this stage (before the autofit) is available as N2Oauto1.pgo.
2. Using the automatic fit results
- Press "Search" in the autofit window- after a short
while, the window will list, for the best fits found:
- nOK - the number of check transitions within
the "Accept" window
- Residual - the RMS observed - calculated for
these check transitions.
- SumI - the sum of observed intensity for these
check transitions.
- The values of the constants obtained for each fit.
- Trial - The number of the trial. (This is
typically only useful for debugging purposes.)
- nDiff - the number of transitions different to
the selected fit. This is only displayed if one of the
fits is selected.
Some additional information is shown in
the
log window.
-
To try out an individual fit, double click
on that row. This will update the line list window with all
the assignments made by that fit, and update the main plot
with a simulation using these parameters. The standard
residuals window will be shown. To try a different one, double
click on that row; all the assignments and parameters will be
updated. The Reset button will remove any
assignments made by the autofit, and reset the parameters to
their initial values.
-
The MinCheck setting deserves
special consideration, as it determines the trials
displayed. The trials are sorted by nOK and then SumI,
with the best fit at the top. If MinCheck is not
zero this sort order is modified; the intent is that it can
indicate how many check transitions are likely not to be
found. If it is set to a positive value then the nOK
is ignored for sorting, provided it is greater than or equal
to MinCheck. A negative value has the same effect,
but the threshold is the number of check transitions + MinCheck.
The default of -1 implies trials with 0 or 1 unsatisfied
check transitions are sorted together. MinCheck
can be changed without re-running the autofit, and the
displayed trials will update accordingly. For the current
case, a value of -1 is allowing too many failed check
transitions, and a value of 0 is more appropriate.
-
For the current case several of the trial
fits with nOK = 4 look good, reflecting the fact
that the P and R branch lines are approximately equally.01
spaced, and several fits with the same pattern of assignments
but with all the J values shifted by the same amount
will give good fits. For this particular spectrum, the best
diagnostic is in fact the position of the Q branch - for the
auto fit run exactly as above only fit number 4 has the Q
branch origin in the right place. (It requires MinCheck =
0 to display.)
- If none of the trials look good, and you want to re-run the
autofit with different settings, the Reset button will
remove any assignments made by the autofit, and reset the
parameters to their initial values.
3. Specifying possible ranges for parameters
While the above process has produced the correct
assignment as one of the possible trials, it is not obvious from
the list presented. The autofit can be made more selective by
specifying allowed ranges for one or more of the parameters to be
determined. This can also speed up the search, as trials are
discarded more quickly. For example, for the spectrum given here
the manual estimate of the band origin is likely to be within 0.5
cm-1 of the actual value. To re-run the search above
with this constraint:
- If you have not done so already, press the Reset button
to discard the autofit assignments
-
To limit the range of a particular
parameter, set the maximum permitted change (+ or -) as the "Std
Dev" for the parameter in the constants window. In this
case set "Std Dev" for the excited state origin to
0.5. Tip: this should be blank if you don't want to apply a
constraint; before autofitting make sure that the standard
deviations of all unconstrained floated parameters are blank,
and don't have standard deviations from previous fits present.
(Click on the down arrow in the autofit window and select "Clear
Parameter Ranges" to do this.)
In the current case, re-running the search with
a constraint of 0.5 cm-1 on the origin promotes the
correct trial to the top entry with nOK = 4, as opposed to 4th.
The percentage of trials rejected is displayed in the autofit
window, and in this case is 90%, as opposed to 1% without the
constraint. Inspecting the rotational constants in the trials
presented suggests a constraint on these might also be
appropriate, perhaps 0.05 cm-1. To apply this:
- Press the Reset button to discard the autofit.
- Set "Std Dev" for B in both the ground and excited
state to 0.05.
This increases the number of failed trials to more than 99.9%.
Once you have found an initial fit that looks
good, press fit a couple of times. This is required as the
parameters would otherwise be the result of a fit excluding any
assigned check transitions. The fit is otherwise normal. The
result at this stage is saved as
N2Oauto2.pgo.
C. Adding More Lines
1. Adding lines that are now (more or less) correctly simulated
- Now add some more candidate lines for assignment in the
region:
-
Open the transitions window "View,
Transitions", and select some lines, say the P and R
branch lines by setting "Change" to "<>",
implying not equal. Some additional selectivity can help -
click on "Filter" to make some extra options
available. The options available are:
-
When you are happy with the selection
displayed click "Add". This will add entries to the
line list window for all the transitions selected by the
window.
- The expectation is that at this stage many lines will be
simulated in approximately the right place. There are two
possible approaches to take, either just taking the nearest
(typically the easiest):
- Select all lines in the line list window (Press All).
- Press the "Nearest" button in the line list
window, which simply assigns each unassigned line to the
nearest line within the acceptance window.
or a modified autofit:
- Select all lines in the line list window (Press All).
- Mark all the lines as search transitions (right click
and select "Mark for Autofit".
- In the autofit window, reduce the search window to, say,
the same as the acceptance window in the line list window,
or something close to the average error in the current
fit. In this case, try 0.01. (The file at this stage is
saved as N2Oauto3.pgo.).
- "Search" will now try all unassigned lines
close to the predicted lines; this should be quick,
provided the window is set small enough. The search in
this case will typically fail for some lines, in which
case only one or two candidate sets of assignments are
produced:
- The upper row takes the closest observed line to each
prediction provided the observed line is within the
search window.
- The lower row simply takes the closest line to each
prediction, regardless of how far out the prediction is.
This is omitted if it would be identical to the upper
row.
In the current case, either produces method gives good
results, so we proceed assuming the "Nearest" method
has been used. The file at this stage is saved as N2Oauto4.pgo.
2. Adjusting assignments using the residuals window
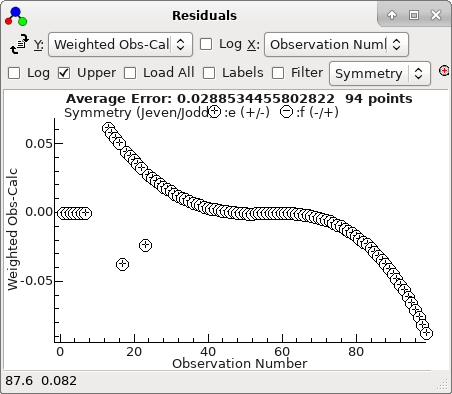
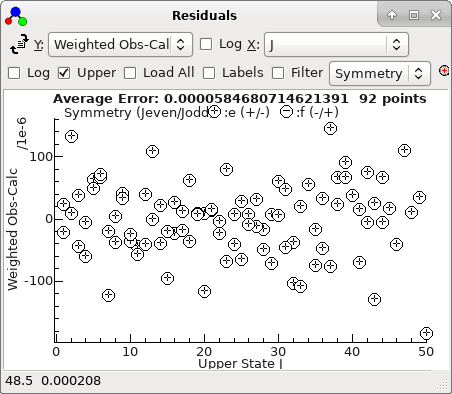
-
The residuals window shows essentially a
regular pattern, with some glitches:
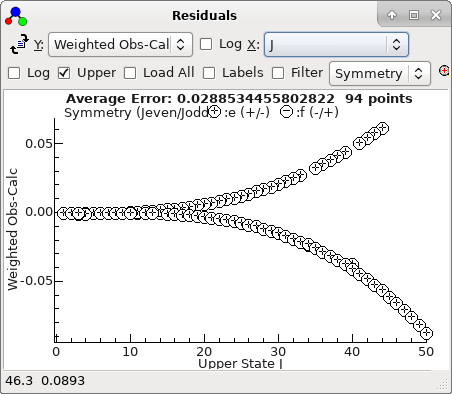
This plot makes more sense if plotted against
J,
rather than observation number:
-
A fit suggests there are two mis-assigned
lines:
-
To fix the mis-assignment right click on
one of the points in the observations window and try one of
the following:
-
Select "
Show and Edit". This
will highlight the relevant observation in the line list
window, and centre the plot on the transition. (This is most
useful if the "
Expand" button (

) is
pressed a few times so the window only shows a small plot
range.)
Here, the wrong choice of line has been made; right clicking
and dragging across the strong line will update the
assignment to the correct value. At this stage, consider
checking suspect or missing lines against the original
spectrum, rather than the line list.
-
The quick fix (... to sweep it under the
carpet) is simply to select Clear Point(s)" on
right clicking on the suspect point in the residuals window.
This will set the "Std Dev" to 0 for the transition,
excluding it from the fit.
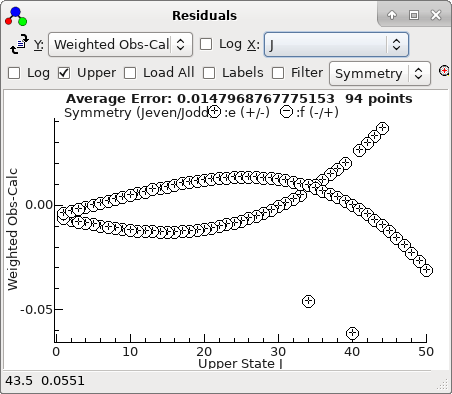
-
With the two bad points removed, the
residuals look good, and can be much improved by floating
D
in both states. refitting gives a good set of residuals saved
as
N2Oauto5.pgo.) The
residuals now look entirely random:
-
To scan for additional assignments after
fitting, on the assumption that some calculated lines will
have moved into the acceptance window:
-
Update the calculated positions selecting
all the lines in the line window and pressing "Update".
This will change the calculated frequency and intensity of
unassigned lines.
-
Press the "Nearest" button. (In
this case it will assign the two peaks removed above.)
- Fit as usual to refine the parameters
The assignment of the P and R branches is now complete; the file at
this stage is saved as N2Oauto6.pgo.
D. Determining additional parameter(s) - the Q branch
In the current case the Q branch is not
simulated, and it needs the determination of an independent
parameter (q). Again this case is sufficiently
straightforward that the assignment could be done manually, but
the autofit process is demonstrated here also. As a single
parameter is to be determined, a single trial fit transition is
required, yielding a very quick search. A possible process is as
follows:
-
Zoom in to the very start of the Q branch,
and right click on a few transitions to add them to the line
list window.
- Select one of these as the trial fit transition, and the rest
as check transitions.
- In the constants window, float q in the upper state.
-
Make sure all all the standard deviations
are blank ("Fix", "Clear All Std Dev...", "Mixture"
in the constants window) otherwise they will act as
constraints on the search.
- A much smaller search window is needed - 0.2 cm-1
should be plenty.
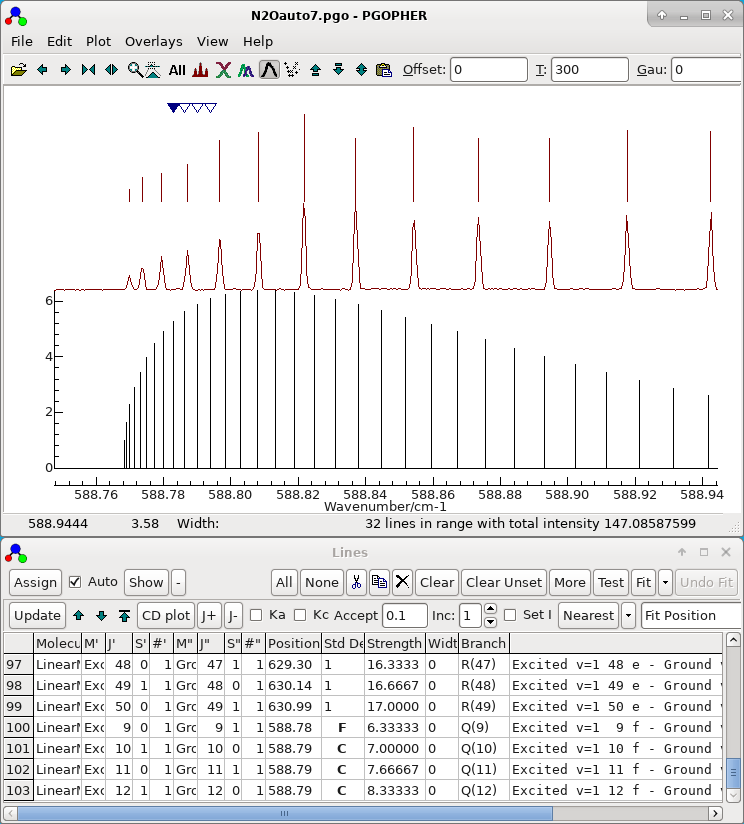
- Note all the parameters determined so far are left floating,
and all the observations assigned so far are left in the autofit
process. The file set up for autofit in this way is saved as N2Oauto7.pgo and the display
should look something like this:
-
In this case the top fit is much better
than the others. The process described in the previous section
under "Adding More Lines" can then be used to assign the
remainder of the transitions. In the transitions window use
set "
Change" to Q) and the "
Nearest" button
will assign almost all of the transitions. After adjusting one
bad assignment, and allowing
qD to
float yields the final result,
N2Oauto8.pgo.
The final fit yields:
SVD fit: 144 Observations, 7 Parameters (scaled)
Initial Average Error: 0.0000586651493283788
Predicted New Error: 0.0000586651493283779
Parameters:
# Old New Std Dev Change/Std Sens Summary Name
1 .4190113492580969 .4190113492579454 2.8034e-7 0.0000 7.7e-10 0.419011349(280) Ground v=0 B
2 1.76173067092025e-7 1.76173067018370e-7 9.963e-11 0.0000 4.2e-13 1.761731(996)e-7 Ground v=0 D
3 588.7679935134044 588.7679935134028 9.3805e-6 0.0000 8.38e-7 588.7679935(94) Excited v=1 Origin
4 .4195742866345234 .4195742866343714 2.7720e-7 0.0000 7.6e-10 0.419574287(277) Excited v=1 B
5 -.00079196442418883 -.00079196442412925 3.2019e-8 0.0000 1.52e-9 -7.91964(32)e-4 Excited v=1 q
6 1.78921147979445e-7 1.78921147906146e-7 9.669e-11 0.0000 4.1e-13 1.789211(967)e-7 Excited v=1 D
7 9.9216205147776e-10 9.9216201696816e-10 1.759e-11 0.0000 8.2e-13 9.92(18)e-10 Excited v=1 qD
Correlation Matrix
Largest off-diagonal element = 0.996 at 4,1 = Excited v=1 B, Ground v=0 B
1 2 3 4 5 6 7
1 1.000
2 0.912 1.000
3 0.102 0.135 1.000
4 0.996 0.901 0.039 1.000
5 -0.151 -0.220 -0.060 -0.161 1.000
6 0.914 0.994 0.072 0.912 -0.234 1.000
7 0.209 0.307 0.084 0.214 -0.954 0.316 1.000
 Procedures Automatic Fitting
Procedures Automatic Fitting