 Procedures Details
Procedures Details | Procedures Details |
<Prev Next> |
PGOPHER can make use of linelists
produced in the ExoMol linelist format, part of the ExoMol project. The way these
linelists are structured make it possible to use many of the
features of PGOPHER on energy levels and transitions
calculated by other programs, to produce energy levels plots, for
example, in addition to simulations. For documentation of the
format see Jonathan Tennyson, Christian Hill, and Sergei N.
Yurchenko AIP Conf. Proc. 1545, 186 (2013) doi:
10.1063/1.4815853.
To produce a simulation first import the .states
file into PGOPHER (File, Import, ExoMol File...).
This will produce a Manifold object (ExoManifold)
containing the states. Then, in the constants window right click
on the Manifold produced and select "Add .trans file...",
and select the required file. The process can be repeated if there
is more than one .trans file. By default, the partition function
is calculated by summing over the energy levels in the .states
file, but the partition function can be specified as a table of
values using an Interpolated
Transition Function. To add this, right click on Molecule,
Add New..., Interpolated Partition Function and then right
click on the Interpolated Partition Function object and
select the file. The resulting file will produce simulations in
the normal way, allowing the temperature and linewidth to be
adjusted and right clicking on any transition will give the
underlying quantum numbers.
The values in ExoMol files can also be used as
input to line position or intensity fitting. To fit to energy
levels, use the procedure for line
position fitting with line lists in separate files, for
which the .states file can typically be used by adding a
few lines to the start of the file. The location of the quantum
numbers and state names in the extra fields after the four
standard ones must be specified by directives in the file as
listed below; the values sn and en are starting
and ending column numbers, with column 1 the first column of the
extra fields. (Omitting en implies all the rest of the
text.) The same procedure can be used to determine the transition
dipole moments by fitting to the Einstein A coefficients
in the .trans file, but the relevant .states
file must be specified with the AddStates directive, and the IntensityUnits in the Simulation must be EinsteinA.
| StateCols s1
e1 [ss] s2 e2 ... |
The state name is selected by
concatenating all the values in the columns s1-e1,
s2-e2 ... of the extra fields. Spaces are
removed and the [ss] text is inserted between the quantum
numbers. The [ss] is optional, and the default separator is
"_", so a set of (vibrational) quantum numbers "0 1 3" would
be converted to a state name 0_1_3. |
| QnoCols s1 e1 s2 e2 ... | The rotational quantum numbers (apart from J,
which is one of the standard fields) are taken from the
given columns. The quantum numbers are assumed to follow the
simple format. |
| AddStates filename |
In .trans files only: read the
energy level definition from the given file. |
| SymCols s1
e1 |
The state symmetry is given in columns s1-e1.
This currently only applies to linear molecules, where the
specified columns should contain the parity (+/- or e/f); it
can be omitted if not required. |
The Include directive, and many others described in Line List Input Format can
also be used.
As an example, the ExoMol SiO linelist (http://www.exomol.com/data/molecules/SiO)
can be imported directly. At the time of writing, three files are
available on the website for the main isotope, 2816for.states,
2816for.trans
and 28Si16O-q.txt
(the partition function) and these can be imported as is using the
procedure above. Importing just the .states file allows
an energy level plot to be produced (View, Levels):
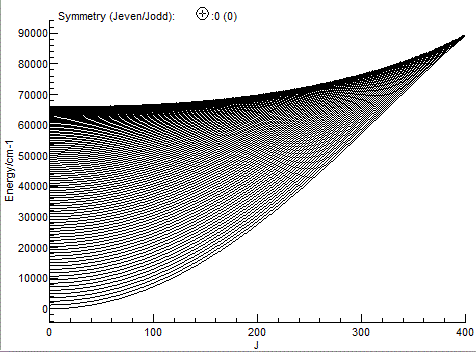
 The file SiOsmall.pgo is a standard PGOPHER
linear molecule file, set up to simulate the v=0 and 1 levels, and
the transitions between them. The partition function is specified
numerically by importing the 28Si16O-q.txt
mentioned above, so that the line intensities are correct at
higher temperatures. The constants were determined by
fitting to the ExoMol energy levels. To determine the rotational
constants (Origin, B D and H) the
following .states file was used as input:
The file SiOsmall.pgo is a standard PGOPHER
linear molecule file, set up to simulate the v=0 and 1 levels, and
the transitions between them. The partition function is specified
numerically by importing the 28Si16O-q.txt
mentioned above, so that the line intensities are correct at
higher temperatures. The constants were determined by
fitting to the ExoMol energy levels. To determine the rotational
constants (Origin, B D and H) the
following .states file was used as input:
Select J" < 100
StateCols 1
1 0.000000 1 0 0
2 1.448467 3 1 0
3 4.345384 5 2 0
... 800 lines omitted
This is the .states file as above, with the lines
referring to the higher vibrational states removed. Note the use
of two directives; the Select J" < 100 is one of the
standard ones, and is used
here to limit the J range of the levels included in the
fit as higher J values fit poorly to the standard centrifugal
distortion expression. The StateCols 1directive is one
described above, and indicates that all the additional quantum
numbers are used to provide the state name. For this to work, the
vibrational states have to be named "0" and "1".
To determine the three transition dipole moments
(0-0, 1-1 and 1-0) the following small .trans file was
used as input:
AddStates 2816small.states
Select J" < 10
StateCols 1
Include 2816small.trans
This file includes two other files - note the use
of the AddState directive to specify the states; the
file 2816small.states is the file used for the energy
level fit above. The 2816small.trans file is directly
from the ExoMol database; lines not involved in the included .states
file are discarded. The other two directives are as above; J
is limited to 10 here as the J dependence of the dipole
moment is not modelled.