Line Position Fitting
This is the preferred method of determining
constants from an experimental spectrum, and can be used when
lines are reasonably well resolved. Where this is not the case it
may be possible to do contour
fitting, but note that this may not give good results.
The procedure described here works best using a
mouse; it is possible with a trackpad or other pointing device,
but a little fiddly. Where right click is required, mac users will
probably want to use Ctrl+Normal Click which is equivalent to
right click for a single button mouse or trackpad.
- Overlay the experimental spectrum
over the PGOPHER
simulation. Note that for the process described here,
peaks in the experimental spectrum should be pointing upwards,
rather than downwards as is sometimes used for absorption
spectra. If the experimental spectrum is plotted separately you
can right click on the experimental spectrum and select invert
or otherwise select Overlays,
Invert from the main menu.
- Drag the simulation
and/or manually adjust the constants so that you can match at
least some peaks between the two spectra. The assignment process
works best if you start with low J transitions near the band origin. To adjust
constants open the constants window
which allows typing in values or adjusting them with the
mouse.
- Right click on a peak that can be assigned to a peak in the
experimental spectrum. A Line List
Window will appear:
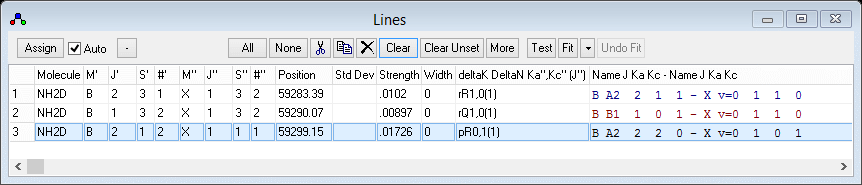
You will normally want to arrange the windows so that the
simulation and the line list do not overlap. Selecting View,
Arrange Windows from the main menu may help.
- All the lines close to the mouse (within a few pixels on the
screen) will be shown selected, which will often catch more than
one rotational transition as above. The intensity of the
individual transitions (shown under Strength) is
normally enough to indicate if you have a sensible selection of
lines. The next step will ignore the weaker lines, or you can
adjust the selected lines with the mouse. (You can delete the
unwanted transitions by selecting the required rows with the
mouse and pressing the delete button
 . Note you can adjust that
the settings controlling the number of peak displayed and
assigned from the Line List Options
Window.)
. Note you can adjust that
the settings controlling the number of peak displayed and
assigned from the Line List Options
Window.)
- Measure the corresponding peak in the experimental spectrum by
dragging the across the peak with the right mouse button. (Press
with the right mouse button to the left of the peak, drag the
mouse to the right of the peak, and release). The peak position
will be determined from the spectrum and a marker indicating the
determined position will be displayed. Note:
- The exact positions you click and release is not important,
provided you do not include any other peak in the range. For
closely spaced peaks you may need to try more than once,
possibly expanding the scale.
- You can remove the markers from the simulation from the
Overlays menu - select "Delete Last Peak" or "Clear All Peaks"
as required.
- Provided Auto is
selected in the line list window, the "Position" column in the
line window of the selected rows will change to that of the
experimental peak and the “Std Dev” will be set to 1
to indicate a measured, rather than a calculated frequency. (If
Auto is off press the Assign
button to do this manually.)
- Repeat steps 3 to 6 to assign a few more peaks.
- To check that you have the correct assignments, press the
"Test" button. The program will read and plot your assignments:
- The observed and calculated values are joined with lines
above the experimental spectrum.
- The upper end of the line is the experimental frequency, and
the lower end is the calculated frequency.
- When the fit is perfect, these will appear as small vertical
tick marks. At this stage the lines will probably be sloping,
but it is normally obvious if you have misassigned something.
- Positioning the mouse over the top of one of the marks will
show the number in the line list of the assignment in the
status bar below the simulation.
- Right click on the mark to show the row containing the
experimental line position in the line list window.
- It is easy to misassign peaks, and wrongly assigned peaks can
distort the fit quite badly. To correct an assignment, select
the corresponding rows(s) with the mouse and either:
- Delete those rows with the delete button
- Re-measure the peaks as in step 5 by dragging across the
peak with the right mouse button.
- Manually edit the values in the grid. You can temporarily
remove an observation by setting the "Std Dev" to zero.
- Given a reasonable number of assigned peaks you can perform a
fit. Select the parameters you want to float by going to the constants window (View,
Constants) and set "Float" to "yes" by each of the parameters
you want to float. Notes:
- You can change between "no" to "yes" by clicking on the
cell, or simply enter "y" or "n".
- It is important to float a small number of parameters to
start with - say only the most significant ones such as the
origin, spin orbit coupling constant, A, (if appropriate) and
rotational constant, B.
- Click Fit in the line
list window.
- A list of observed and calculated transition frequencies,
the average overall error, new values for the constants and
estimates for their uncertainties and a correlation matrix
will be displayed in the log window
(View, Log though this will normally show automatically. You
will normally want to arrange your desktop so that the
simulation, line list and log windows do not overlap ). For
more details on the values displayed, see the documentation
for the log window.
- A plot of the residuals before the fit will be shown in the
residuals window. This is
normally shown automatically, or use View, Residuals.
- The main window will show an updated simulation (unless you
have AutoReplot turned
off, in which case you will need to press the "Simulate"
button).
- If you used an offset on the simulated spectrum you will
probably need to reset the offset to zero after the first fit.
- If the results look reasonable, then press "Fit" repeatedly
until the error converges. Note that the residuals and
calculated positions displayed are the values before the fit,
rather than after.
- If the fit does not look good, the Undo Fit
 button in
the log window will step constants back one fit cycle for each
press
button in
the log window will step constants back one fit cycle for each
press
- If you go too far, the Redo
Fit
 button in the log window will step
the constants forward one fit cycle.
button in the log window will step
the constants forward one fit cycle.
- To correct assignments, see step 9.
- Complete the fit by repeating from step 3, adding more peaks
to the line list and floating more parameters.
- A complete report on the fit can be obtained from the Log
window:
- Clear the log window with the clear button

- Press the "fit" button in the line list window
- In the log window the copy button:
 will copy the report
to the clipboard, or alternatively use the save as button
will copy the report
to the clipboard, or alternatively use the save as button  to save the log to a
file.
to save the log to a
file.
- The contents of the line list window are normally saved to,
and loaded from, the .pgo file, though it is more flexible to
save the assignments to a separate file. See the next section
for how to do this.
Common Problems
Line lists in
separate files
It is normally more flexible to work with assigned lines in an
external file for larger fits as only limited editing is
possible in the line list window. External files are text files,
normally with a .lin or .obs extension, and will accept a wide
variety of directives and formats in addition to the line list
as displayed in the line window. Directives allow more control
over the fit; for example, a "stop" directive will cause the rest of the
file to be ignored, allowing fits to part of the data, and a "Units"
directive allows the units to be set for the following lines
allowing, say, data from microwave and infra-red spectra to be
combined. For more details see Combined
and Multi-State Fits and the Line
List Input File Format. A custom text editor, the Tabslave Editor, is distributed
with PGOPHER which
has been specially designed for editing these files. See Fitting example - the A-X transition in C3
for a worked example in line position fitting involving two
overlapping transitions, which demonstrates the process, and why
it might be useful. The procedure below describes how to
generate a basic version of the file from the line list window.
- Press
 and select "Save to File"
to save the current line list to a file. Any of the formats
presented will work in simple cases; see the discussion in Multiple State Fitting example - the A-X
transition in C3 for more information on the
possibilities. Use "Branch" or "Simple" format
for straightforward applications.
and select "Save to File"
to save the current line list to a file. Any of the formats
presented will work in simple cases; see the discussion in Multiple State Fitting example - the A-X
transition in C3 for more information on the
possibilities. Use "Branch" or "Simple" format
for straightforward applications.
or (to set a previously generated file):
- Go to the log window(View, Log).
- Press the
 button and
browse to the file (or where you would like the file to be
created). This will put the file name in the observations file
name window. (The file name should typically end in .lin or
.obs).
button and
browse to the file (or where you would like the file to be
created). This will put the file name in the observations file
name window. (The file name should typically end in .lin or
.obs).
- Press the Edit Button
 . If the file does not
exist you will be asked if you want to create it. If the Slave Editor is set up correctly,
this should start.
. If the file does not
exist you will be asked if you want to create it. If the Slave Editor is set up correctly,
this should start.
To add observations to the file:
- Use "More, Copy Branch Table", "More,
Copy Simple Format" or the copy button(
 in the line list window)to
put your assignments onto the clipboard. These produce different
text formats, and one of the first two is likely to be easier to
work with.
in the line list window)to
put your assignments onto the clipboard. These produce different
text formats, and one of the first two is likely to be easier to
work with.
- Paste the assignments into your observations (.lin) file, and
save it.
To use this file, rather than
lines in the line position window:
- Use the Fit Button
 in the log window, rather
than the line list window. If the Slave
Editor is in use, then the contents of all slave editors
are saved before starting the fit. Note that if a slave editor
is unable to save a file that has been modified then the fit
will not start and the slave window will come to the front or
flash.
in the log window, rather
than the line list window. If the Slave
Editor is in use, then the contents of all slave editors
are saved before starting the fit. Note that if a slave editor
is unable to save a file that has been modified then the fit
will not start and the slave window will come to the front or
flash.
- Assignments are best added to the
external file by copying and pasting from the line list window.
 Procedures
Procedures