 Windows
Windows
| Windows
|
<Prev Next> |
This window displays the position, assignment and intensity of lines in the simulated spectrum. It appears when right-clicking on a peak in a simulated spectrum, and on each click all the lines close to the clicked position are added to this window, with the most intense first. You can adjust the number of lines added from the Line List Options Window. Simple line position fits can also be done directly from this window, though for anything other than the simplest of fits more the line lists are best placed in a text file as only limited editing is possible in the this window. (An initial text file can be created by copying and pasting from the line list window.) The contents of this window are normally (for version 5.2 and above) saved to and loaded from the .pgo file.
Click on an item in the picture below for more information: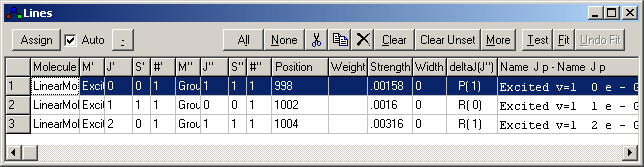
 |
Set
frequency of selected lines
from clipboard, assuming a number is available as text on the
clipboard. The weight is also set to 1. Measuring a peak by right
clicking and dragging
over a peak in an experimental or simulated spectrum in PGOPHER puts the peak position
on the clipboard for this purpose. If the neighboring "Auto" item is
checked (the default) this assignment is done automatically each time a
peak in an experimental spectrum is measured. A blue line in the
experimental spectrum will appear, indicating the measured peak
position. |
 |
If on, assign measured peak to selected transitions automatically (if off, this can be done manually using Assign). |
 |
Show difference between first and last line selected. |
 |
Select all lines in the line list window. |
 |
Select no lines. |
 |
Copy
selected lines to clipboard
and delete them. |
 |
Copy selected lines to clipboard. |
 |
Delete selected lines. |
| Delete all lines. | |
 |
Delete unassigned lines, i.e.
those with the weight column empty. |
 |
Menu for more actions:
|
 |
Plot assignments and display residuals |
 |
Fit observations in this window |
 |
Reset parameters to the value
before the last fit cycle. This button is disabled until a fit has been
done. |
| Molecule | The molecule responsible for this transition. |
| M' | Manifold for the upper state of the transition |
| J' | Upper state total angular momentum or M'. (M' in the presence of static field,
otherwise J' in the absence
of hyperfine
structure, otherwise F'). |
| S' | Symmetry of the upper state. |
| #' | Eigenvalue number for upper state. This gives the index (starting from 1) of the upper energy level with respect to other levels of the same total angular momentum and symmetry. |
| M'' | Manifold for the lower state of the transition |
| J'' | Lower state total angular momentum or M". (M" in the presence of static field, otherwise J' in the absence of hyperfine structure, otherwise F'). |
| S'' | Symmetry of the lower state. |
| #'' | Eigenvalue number for lower state. This gives the index (starting from 1) of the upper energy level with respect to other levels of the same total angular momentum and symmetry. |
| Position | The position of the transition; the units for this are the
units used in the main simulation window. For line position fitting,
alter this number to the position of the observed peak, either manually
or (more usually) with the assignment process described under line
position fits. |
| Weight | The weight of this transition in a line
position fit. If this is negative, zero or blank this transition
will not be included in the fit. It is initially blank, and the
automatic assignment process will set this to 1. The weight is formally
the standard deviation of the measured peak position, so less certain
measurements should be given larger values. Note that the parameters
produced by a fit only depend on the relative values of the weights. |
| Strength | The intensity of the rotational transition. |
| Width | The width of the transition. (Note that this does not include
the Gaussian or Lorentzian width set on the plot window.) |
| deltaJ(J'') | The transition label in branch notation, such as P(1). The
display will depend on the molecule type and the electronic and nuclear
angular momenta included in the calculation. |
| Name | Details of the transition, including other quantum numbers
where appropriate. The assignment process will also add the source of
the measurement (typically a filename) where possible. |