 Molecule Types Vibrational Structure Force Field Analysis
Molecule Types Vibrational Structure Force Field Analysis | Molecule Types Vibrational Structure Force Field Analysis |
<Prev Next> |
The z matrix object is used to specify
the the geometry of the molecule using the z matrix format
commonly used by ab initio programs or standard Cartesian
coordinates (or a mixture of the two). The units used are atomic
mass units, Angstroms and degrees, independent of the units
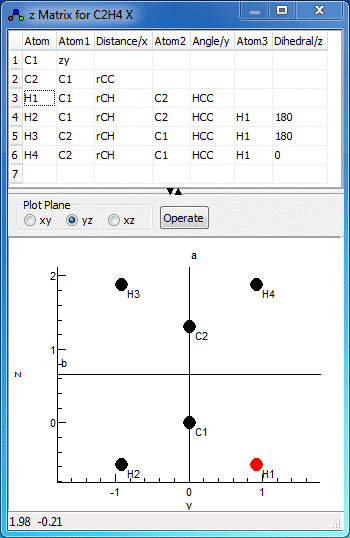
setting in the simulation object. The z matrix window, shown below
for C2H4, allows the display and editing of
this geometry. Provided the z matrix object is set active,
changes in this window will be reflected in the Nuclear co-ordinates objects. To
create a z matrix object, right click on the Electronic State object
object and select "Add New..., z Matrix". To bring up the
window right click on the z matrix object and select "View...".

| Check |
Check the contents of the z matrix
grid, and update the plot to match. Note that the Nucleus objects for the
state are not updated immediately by this - Apply to State
(below) will force this, or running a full calculation if
the z matrix is active. |
| Print |
Check the z matrix, and display
information on the geometry, moments of inertia and
principal axes in the Log Window. |
| Apply to
State |
Update the Nucleus
objects for the state to reflect the current z
matrix. This is only required if the Active
property of the z matrix is false. |
| Load from
State |
Clear the z matrix, and set from the
Nucleus objects for the
state. This will use Cartesian format. |

| Active | Set true to use this z matrix for geometry, in
which case the Nuclear
co-ordinates objects are updated from the z
matyix. There must be no more than one active z
matrix at a time. |
| AverageMass | If true, use the average mass of a nucleus if the isotope
is not specified. If false, the exact mass of the most
abundant isotope is used. |
The format required for the z matrix is as used
by many ab initio programs, and elsewhere, and it should
be possible to copy the z matrix to and from other programs with
minimal or no editing.
The even numbered columns allow up to three
connected atoms to be specified; the name used should appear in
the first column of one of the rows above. If these are all blank,
As a special case, on the first line where no linked atom can be
specified, the second column (if not blank) is taken as specifying
the plane to place the first three atoms in. The first letter
specifies the axis on which the first two atoms lie, and the
second character (if present) completes the specification of the
plane.
The odd numbered columns specify a distance (in Angstroms) and up to two angles (in degrees). The first angle (in column 5) is required after the second atom specifies the angle between the first three atoms on the line. For atoms after the third a dihedral angle must also be specified, which gives the angle between the planes containing the first three and last three atoms
The bond lengths and angles can be given as
simple numerical values, but can also be given as general
expressions involving symbolic values. This is typically more
useful, as it allows symmetry constraints to be added naturally.
For the C2H4 example above, note the use of
two bond length variables (rCC and rCH) and a bond length variable
(HCC). These can be used in expressions if required - an
expression such as 180-HCC/2 might be needed in other
contexts. When scanning the z matrix, unknown variables are
automatically created as parameters to the current z matrix
object, and they can then be edited in the Constants Window. If dealing with
more than one isotopologue, these variables are better placed in a
variables object, (with Global set to true) so
the same value can be used in all species.
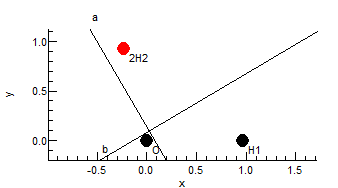
If all the linked atoms in a row are blank, then Cartesian format
is assumed, and x, y and z should be given
in the odd numbered columns. Cartesian coordinates can be mixed
with