The Jahn-Teller effect in the B1E" state of NH3
This force field analysis includes the Jahn-Teller effect, and
shows how isotope independent potential energy surface parameters
can be used to calculate vibrational parameters and energy levels
in NH3, NH2D, ND2H and ND3.
The work is described in A.K. Saha, G. Sarma, C-H Yang, S. van de
Meerakker, D.H. Parker and C.M. Western, Phys. Chem. Chem. Phys.,
(2015), doi:10.1039/C5CP01299F.
There are 4 files:
- BSimpleHarm.pgo: A
simple force field analysis for the B1E" state of NH3,
ignoring Jahn-Teller effects. This is used to produce table 4 in
the paper.
- BSimpleHarm.obs:
The observation file used.
- BharmJT3sym.pgo:
The multi-isotope model, with all 4 isotopologues and 2
electronic states for each.
- BharmJT3sym.obs:
The observation file used to produce table 7 in the paper.
Notes
- Neither .pgo file simulates a spectrum, as only one electronic
state is set up in the file.
- The point group used throughout is C2v, even for NH3
and ND3 which are D3h. One of the
consequences of this is that the modes are numbered 1..6, rather
than the more normal 1..4. In the paper the numbering scheme 1,
2, 3x, 3y, 4x, 4y is used,
corresponding to 1..6.
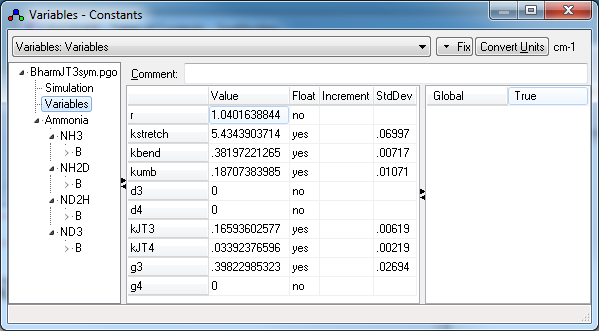
- The structure of the multi-isotopologue file is rather
complex. At the upper levels it contains a variables object with the force
constants, and a molecule object for
each isotopologue, each of which has a single manifold object:
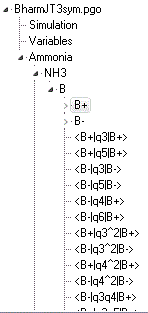
- Each manifold object has two electronic state objects, B+
and B- (|+> and |-> in the paper), and multiple perturbation objects
acting within and between the states. These are calculated from
the force constants in the variables
object controlled by settings in the internal coordinates object.
-
This corresponds to equation (33) in the
paper. Note the the first three terms contribute to
perturbations acting within the B+ state, possibly including <B+|q1|B+>,
<B+|q3|B+> and <B+|q3|B+>,
depending on the isotopologue. The last two correspond to the
second term in equation (33) and contribute to perturbations
acting between the two electronic states, <B-|q4|B+>,
<B-|q6|B+>. The B- states only have the terms
acting within the electronic state, as the terms acting
between states do not need to be duplicated:
Note the sign changes compared to the B+ entry, reflecting the
± in equation (11).
- View, States will show the calculated energy levels -
note that this is quite a time consuming calculation. The
individual vmax entries
have been adjusted to the minimum values converge the
calculation.
 Molecule Types Vibrational Structure Force Field Analysis Samples
Molecule Types Vibrational Structure Force Field Analysis Samples 
