 Molecule Types Vibrational Structure Vibrational Energy Levels and
Franck-Condon Factors
Molecule Types Vibrational Structure Vibrational Energy Levels and
Franck-Condon Factors | Molecule Types Vibrational Structure Vibrational Energy Levels and
Franck-Condon Factors |
<Prev Next> |
ab into programs can be used to calculate the normal mode information required to simulate electronic spectra including Franck-Condon factors. The key parameters required for are summarized in Making a data file for simulating vibrational structure from l matrices. The steps below provide a worked example for the first band in the photoelectron spectrum of water, using ab into calculations for the required values. See the related worked example, The Photoelectron Spectrum of H2O for an alternative approach using literature values.
For this exercise Molpro is used, though many ab initio programs will produce the same information. A calculation including normal modes can be performed with the following control file:
re=1.013
r1=re
r2=re
a=109.41
basis=VDZ
geometry={
ANGSTROM;
H1;
O1,H1,r1;
H2,O1,r2,H1,a;
}
wf,9,2
hf
optg
frequencies
casscf
optg
frequencies
This performs a geometry optimization and normal mode
calculation for the ground state of the ion using both
Hartree-Fock and CASSCF. The same file will also work for the
ground state of the neutral if the "wf,9,2" line is removed. The more recent
versions of Molpro output in XML format that PGOPHER can read
directly; alternatively add a "put molden,H2O.molden" command after the frequencies step. This
writes out the normal mode (and other) information that PGOPHER can also read
directly.
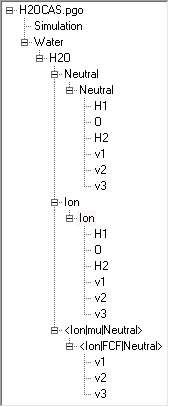
The normal mode output that is required looks like this for the neutral molecule:
FREQUENCIES * CALCULATION OF NORMAL MODES FOR CASSCF000
Atomic Coordinates
Nr Atom Charge X Y Z
1 H1 1.00 0.000000000 -1.417298632 1.034331878
2 O1 8.00 0.000000000 0.000000000 -0.130322946
3 H2 1.00 0.000000000 1.417298632 1.034331878
Frequencies dumped to record 5400.2
Gradient norm at reference geometry: 0.46187D-03
Normal Modes
1 A1 2 A1 3 B2
Wavenumbers [cm-1] 1715.28 3721.36 3833.40
Intensities [km/mol] 53.05 2.68 14.45
Intensities [relative] 100.00 5.06 27.23
H1X1 0.00000 0.00000 0.00000
H1Y1 0.42031 0.56516 0.52478
H1Z1 0.53260 -0.39609 -0.43124
O1X2 0.00000 0.00000 0.00000
O1Y2 0.00000 0.00000 -0.06612
O1Z2 -0.06711 0.04991 0.00000
H2X3 0.00000 0.00000 0.00000
H2Y3 -0.42031 -0.56516 0.52478
H2Z3 0.53260 -0.39609 0.43124
Note the optimized equilibrium geometry (in atomic units), the normal mode frequencies and the l matrix elements at the end. The latter are the relative motion of each atom in a particular mode.
Press the simulate button ( ) and
then the all button (
) and
then the all button ( ) and you should see a
simulation. Comparison with the empirical spectrum generated in
the related worked example, The
Photoelectron Spectrum of H2O shows that the
intensities are reasonable, though both the ionization energy
and vibrational frequencies need adjustment.
) and you should see a
simulation. Comparison with the empirical spectrum generated in
the related worked example, The
Photoelectron Spectrum of H2O shows that the
intensities are reasonable, though both the ionization energy
and vibrational frequencies need adjustment.
The resulting file is available as H2OCAS.pgo.