23 Mar 2022: Regrettably, further updates to PGOPHER will no longer be available.
10 Dec 2020: MAC versions Initial testing on Big Sur (version 11) suggests PGOPHER runs on both Intel and M1 Macs using the Cocoa Development Version, specifically pgopher-MacOSX-x86_64-cocoa.app.zip. Running on M1 machines currently uses Rosetta.
1 Sep 2020: MAC versions Running on 10.15 (Catalina) requires the Development Version, specifically pgopher-MacOSX-x86_64-cocoa.app.zip. The reliability of this version has improved significantly since Oct 2019.
13 Oct 2019: Upgrading to MACOS 10.15 (Catalina) is not recommended as this is not yet fully supported by PGOPHER. Apple no longer supports 32 bit programs, but the required underlying 64 bit library required has only recently become available in a useable state, and some development work is required for this to work with PGOPHER. A 64 bit Mac beta version is available from the Development Versions directory.
5 Jan 2019: Important note on
simulating microwave spectra: if you have quadrupole
structure, check the setting of the AllowComplex
flag. For almost all types of spectra it makes no difference but
setting this to true is required for some cases involving large
hyperfine structure, such as found with iodine. The default is
currently false (which is faster), but the next version will
change this to true as it has become clear that it is too easy to
perform incorrect calculations. The requirement is triggered by
exotic terms in the Hamiltonian, χab, χac
and χbc in the case of iodine, and other uncommon
terms including perturbations may also trigger this requirement.
Thanks to Susanna Stephens for a discussion prompting this change.
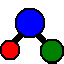
3 Dec 2018: Paper describing updated automatic fitting and Nearest Lines Plots available on-line: “Automatic and semi-automatic assignment and fitting of spectra with PGOPHER”, Colin M Western and Brant E Billinghurst, Physical Chemistry Chemical Physics, 2019, doi:10.1039/c8cp06493h; the accepted manuscript is available here.
Version 10.1.180 (30 Nov 2018): Formal
release of version 10.1. This includes a significant update to the automatic fitting, particularly the addition
of Nearest Lines Plots;
see Automatic fitting of the nu5 band of cis 1,2-Dichloroethene,
Automatic fitting of an N2O IR spectrum
and Automatic fitting of an N2O IR spectrum with additional diagnostic tools
for detailed instructions.
A variety of other improvements have been made, including improvements
in line intensity fitting and better handling of larger problems. See the release notes
for a
more detailed list of changes and bugs fixed. Upgrading is
recommended for all users to make use of speed ups and bug fixes; the
user interface has not changed significantly, apart from some changes to the automatic fitting process,
and the .pgo files from older versions will work in the new version except for unusual cases, which will generate a message on loading.
8 Feb 2018: If EigenSearch is set False for a Manifold (not the default), then under certain circumstances (typically simulating after a fit) spurious eigenvectors can be used for subsequent calculations, which can show up as incorrect intensities. This is fixed in development versions after 10.1.157, or in older versions by making sure EigenSearch is true or invoking "View, Clear Cache" before calculations.
27 Apr 2017: Loading .pgo files from older versions will set the eigenvalue number columns (labelled #) in the line list window to a negative number if they were saved as blank. This will give an error message on fitting; simply clear the relevant entries or use a development version.
4 Apr 2017: Paper describing Automatic Fitting as implemented in version 10.0 now available on line: "Automatic Assignment and Fitting of Spectra with PGOPHER". C. M. Western and B. E. Billinghurst, Physical Chemistry Chemical Physics, 19, 10222 - 10226 (2017), doi:10.1039/c7cp00266a. The accepted manuscript is also available.
Version 10.0.505 (14 Feb 2017): Formal release of version 10. The major new feature is Automatic Fitting as described in "Automatic assignment and fitting of spectra with PGOPHER", but there are many other improvements and fixes, including faster handling of large problems and importing large files with spectra. See the release notes for a more detailed list of changes. As ever, .pgo files from older versions will work without modification in the new version, except in a few unusual cases, which will give a warning message on loading. The .pgo files remain essentially compatible with older versions, though expect warning messages if newer features are used. The doi for version 10.0 is doi:10.5523/bris.160i6ixoo4kir1jxvawfws047m
Version 9.1.100 (8 Jan 2016): Formal release of version 9. In addition to the new features listed below, a comprehensive tutorial Walkthrough of Simulating and Fitting a Simple Spectrum has been added to the documentation, including an N2O sample spectrum to work with, and other new features include Custom Width Functions to provide a simple empirical way of specifying quantum number dependent line widths and intensities and a significant rework of the Transitions Window to allow selection by more quantum numbers and other options. See the release notes for a more detailed list. As ever, .pgo files from older versions will work without modification in the new version, except in a few unusual cases, which will give a warning message on loading. The ..pgo files remain essentially compatible with older versions, though expect warning messages if newer features are used
Version 9.0.101 (5 July 2015): Update of Mac version only, to avoid hard crash on some error messages. Mac users should update. Minor updates to documentation also.
Version 9.0.100 (17 June 2015): Draft release of version 9. New features include faster calculations for larger problems, Loomis-Wood plots, a wider selection of units for calculated quantities (for example Angstroms and Oscillator Strength), a Vibrational Partition Function for calculating complete partition functions, a Check Derivatives command to assess the accuracy of numerical derivatives, other tools to assist in fitting and many other smaller improvements and bug fixes. See the release notes for a more detailed list. .pgo files from older versions will work without modification in the new version, except in a few unusual cases, which will give a warning message on loading. A permanent DOI will be made available for the new version shortly. Bug reports welcome to help-pgopher@bristol.ac.uk.
26 Jan 2015: If simulating hyperfine structure on a molecule with equivalent nuclei, then the statistical weights are not calculated correctly if equivalent spin 0 nuclei are explicitly included in the calculation. This is fixed in version 8.0.308; for earlier versions simply delete any nucleus objects with spin zero. The documentation on how to handle equivalent nuclei when calculating hyperfine structure has also been clarified - see linear and asymmetric top documentation.
20 Nov 2014: Versions before 8.0.258 swapped the +l and -l labels in symmetric tops for levels of E2 and E5 vibronic symmetry (not rovibronic symmetry) as compared to the definition given by Hoy and Mills J. Molec Spectrosc. 46, 333 (1973). The calculated energy levels and intensities are not affected, just the labels of the states. This is fixed in the deveopment versions; note input files for fitting produced by or for older versions may require adjustment for degenerate vibronic states in the affected point groups (D4d and other groups with 5 fold or higher rotation axes).
23 July 2014: In addition to the website, PGOPHER can now be cited via a permanent digital object identifier, doi:10.5523/bris.huflggvpcuc1zvliqed497r2.
23 April 2014: Two bugs have been found concerning simulating Stark and Zeeman effect spectra. Spectra in the presence of external fields involving symmetric tops with degenerate vibronic states included spurious extra transitions, and the M dependence of the intensity of multiphoton transitions, or ordinary transitions with random polarisation, was calculated incorrectly. These have been fixed in development versions above 8.0.186.
Version 8.0.102 (11 December 2013): Minor update to fix crashes in energy level plots in the Mac version, and some issues with vibrational calculations. See the release notes for a more detailed list of problems fixed; there are no new features
- Custom population functions, allowing an essentially arbitrary function to be used as an alternative to the Boltzmann distribution.
- Flexible use of HITRAN and ExoMol linelists.
- Axis switching effects
- Custom transition moment functions to allow for Herman-Wallis factors
- Tweaks for Mac usage, including more (Mac) standard shortcut keys and dropping of files onto main window.
- 64 bit version now part of the standard release,
allowing
larger calculations to be performed.
Introduction
PGOPHER is a general purpose program for simulating and fitting rotational, vibrational and electronic spectra. It represents a distillation of several programs written and used over the past decade or so within the Bristol laser group and elsewhere, but is a re-write from scratch to produce a general purpose and flexible program. PGOPHER will handle linear molecules and symmetric and asymmetric tops, including effects due to unpaired electrons and nuclear spin, with a separate mode for vibrational structure. The program can handle many sorts of transitions, including Raman, multiphoton and forbidden transitions. It can simulate multiple species and states simultaneously, including special effects such as perturbations and state dependent predissociation. Fitting can be to line positions, intensities or band contours.